| Trouble identifying glycan modified peptides and quantifying ions in DIA data | nkegley | 2024-03-08 11:00 | |||||||||||||||||||||||
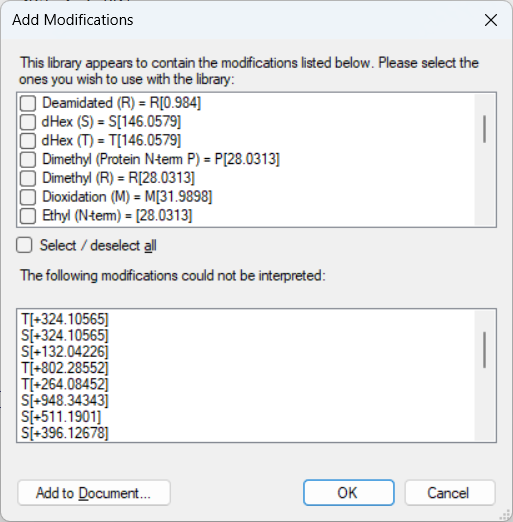
I am attempting to use Skyline to quantify ions from an existing DIA dataset I've generated using our labs orbitrap instrument. When using skyline to build a library using the related DDA dataset and extracting chromatograms using the DIA run, I get an error window about not being able to identify modifications (mostly glycans on Ser and Thr residues). Rebuilding glycan modifications in the "modifications" window of skyline doesn't seem to remedy this, and when I get the library built by skyline, all I generally see are peptides with fixed carbamidomethylation, no glycans. I've used Byonic software to produce an mzident file for use in skyline, and there I can detect PSMs that include the glycan modifications (which makes sense considering I've already built an inclusion list for our instrument for those exact masses). Because of that, I assume I'm just going wrong somewhere in the skyline settings that results in ions from the glycan modified peptides being excluded from the library/chromatogram extraction. Any help or advice would be appreciated! |
|||||||||||||||||||||||||
| |||||||||||||||||||||||||
 AddModificationsDialog.png
AddModificationsDialog.png