| Intensity Threshold on MS2 fragments?? | zrhopkins | 2024-03-14 04:37 | |||||||||||||||||||||||||||||||||||
I was wondering if there is some in the background built-in intensity threshold on whether or not Skyline will show chromatograms for MS2 data? We are using Skyline to quantify data collected in an Thermo Exploris 120. We have found that sometimes Skyline will not show MS2 chromatogram data for a fragment of a compound. However, when we review this data in Thermo software, there is clearly MS2 data generated for the fragment of interest. I have attached an example. Showing 1) how in Skyline we don't see any fragment data for the 98 and 79 m/z and 2) If that same data is viewed in Thermo software I see the fragmentation event and a low intensity peak for the 98 and 79 m/z. Maybe there is something that I'm missing, but any insight would be helpful! |
|||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
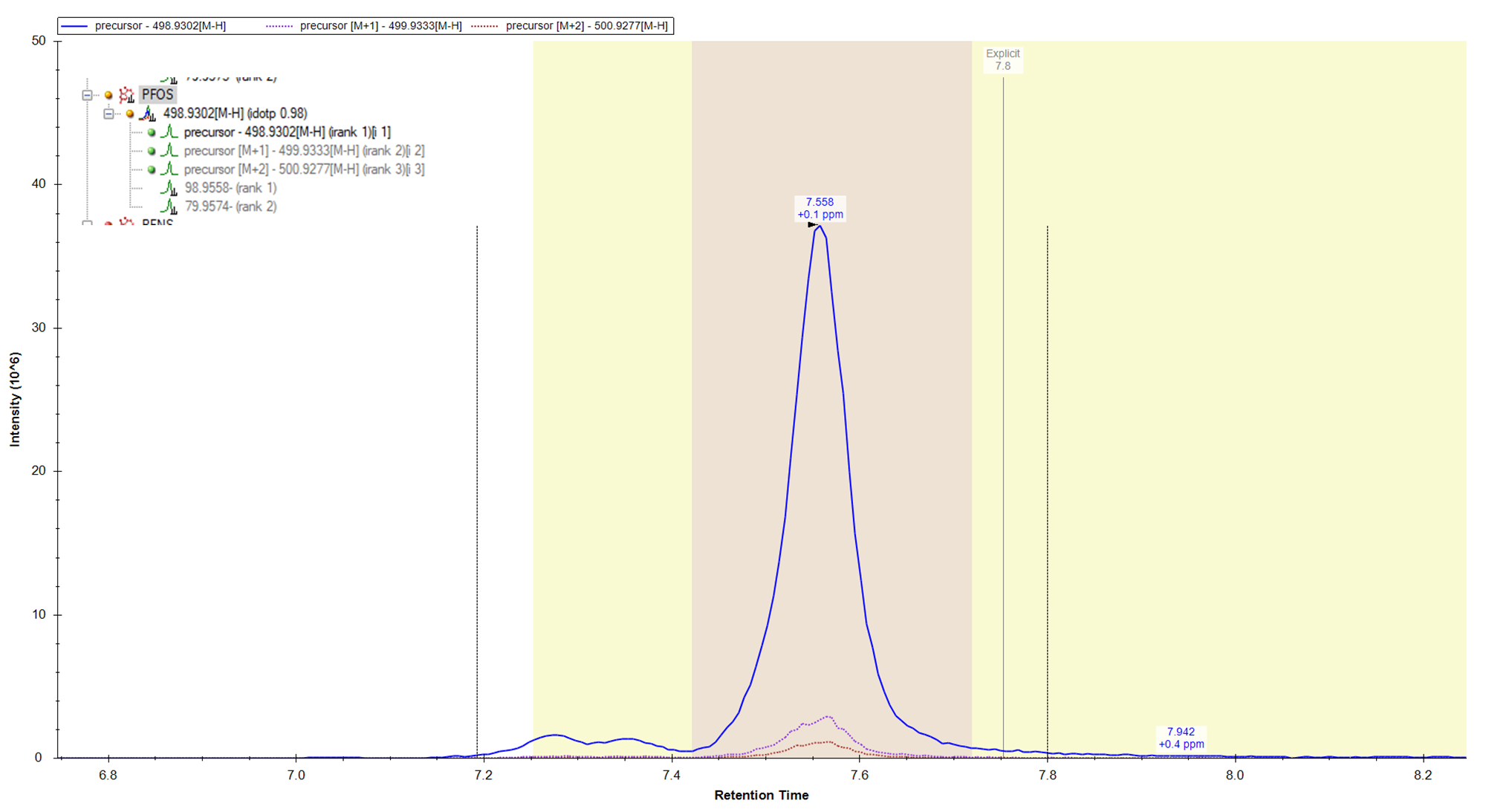 Skyline_Chromatogram.png
Skyline_Chromatogram.png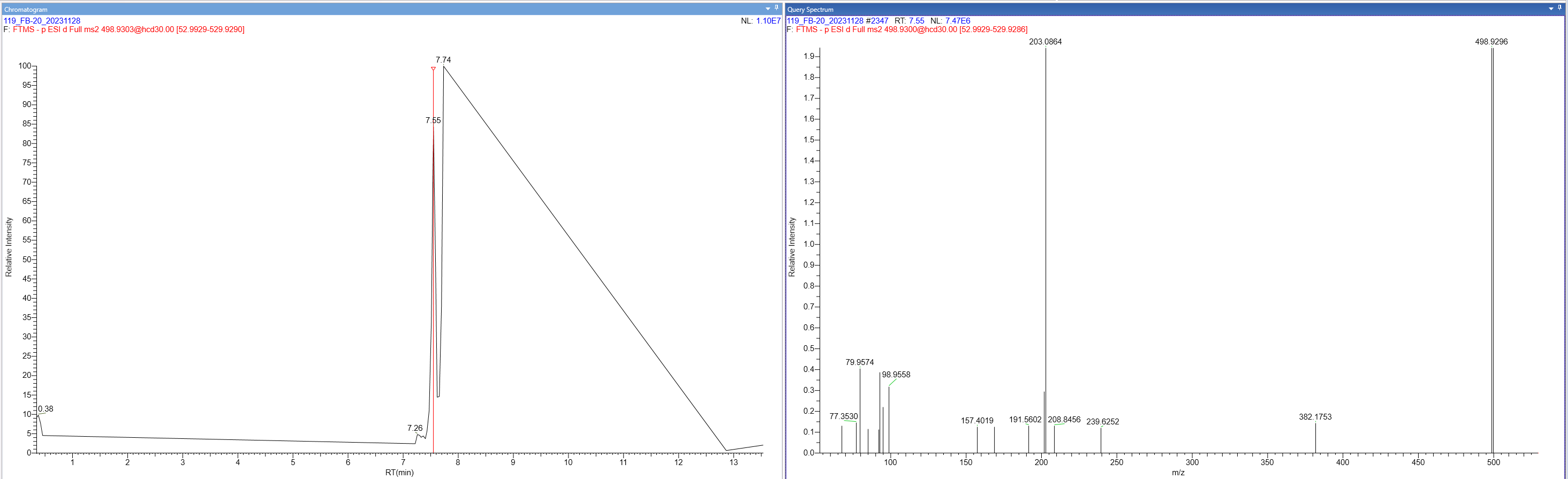 Thermo_MS2_Result.PNG
Thermo_MS2_Result.PNG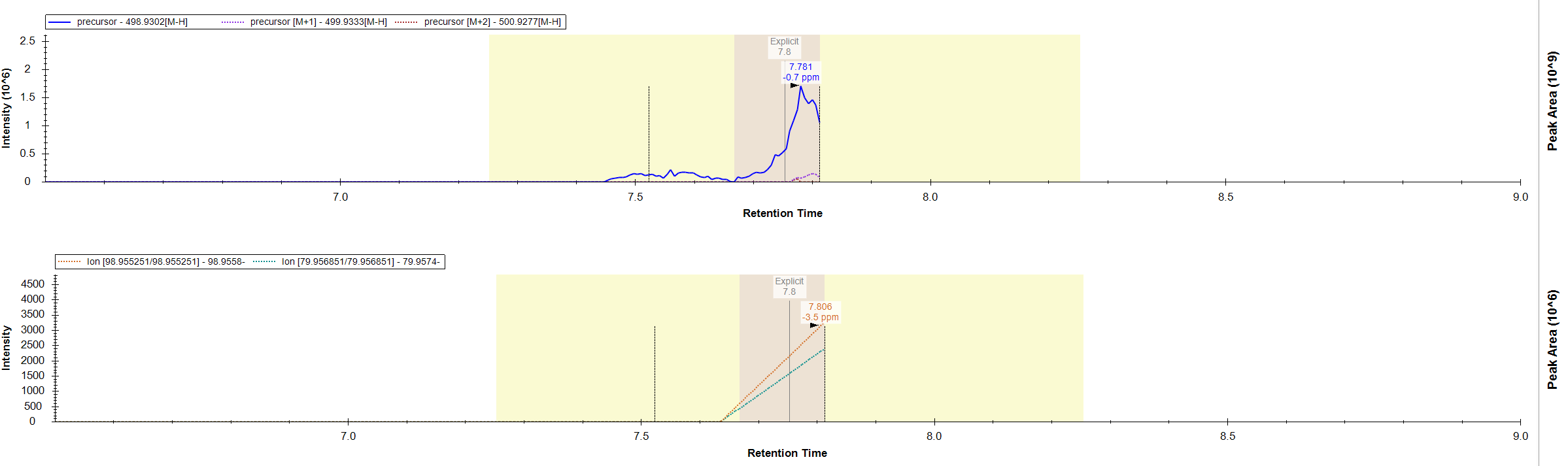 Skyline_PRM_Chromatogram.PNG
Skyline_PRM_Chromatogram.PNG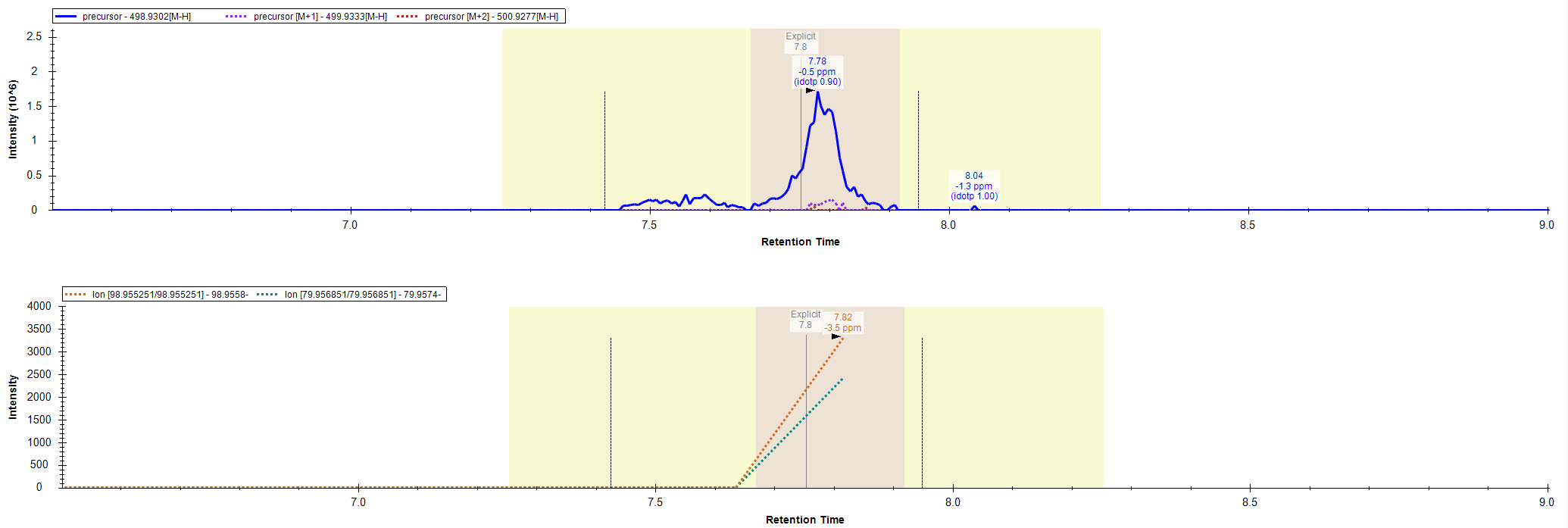 Skyline_DDA_Chromatogram.PNG
Skyline_DDA_Chromatogram.PNG